
Propionacidemie is een zeer zeldzame, erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en de afbraak van stoffen in ons lichaam bedoeld. Dat is nodig voor de opbouw van weefsels, zoals spieren botten en organen en voor het vrijmaken van energie. Deze aanmaak en afbraak verlopen in verschillende aparte stappen. Voor elke stap is er meestal een specifiek enzym dat ervoor zorgt dat de stap kan doorgaan. Als zo’n enzym niet goed werkt, biochemische afwijking, blokkeert de stofwisseling op die stap, wat aanleiding kan geven tot een stofwisselingsziekte.
Bij patiënten met propionacidemie is het enzym propionyl-CoA carboxylase niet, of minder goed werkzaam. Daardoor kunnen de aminozuren valine en isoleucine niet goed door het lichaam van propionacidemie-patiënten worden verwerkt. Als de stofwisseling van kinderen met propionacidemie teveel valine en isoleucine aangeboden krijgt, ontstaat er een stijging van propionzuur, wat schadelijk is voor verschillende organen en voor de hersenen. Aminozuren zijn de bouwstenen van eiwitten. Eiwitrijke voeding, zoals vlees, vis, zuivelproducten bevatten dus veel valine en isoleucine.
Propionacidemie is een zeer zeldzame stofwisselingsziekte, die voorkomt bij 1 op de 100.000 pasgeborenen in Vlaanderen.
Diagnose
De diagnose wordt gesteld op het acylcarnitineprofiel (verhoogd C3-acylcarnitine) op gedroogd bloed en door meting van de organische zuren in urine. DNA-analyse zorgt voor aantonen van het precieze defecte gen. Eventueel kan bij twijfel de restactiviteit van het enzym gemeten worden in witte bloedcellen, maar meestal zijn daarvoor fibroblasten nodig (stukje huid dient dan afgenomen te worden).
Het acylcarnitineprofiel in een gedroogd bloedspotje geeft de diagnose in stabiele omstandigheden in de neonatale periode, en tijdens decompensatie op latere leeftijd. De techniek die hiervoor aangewend wordt is de tandem-massaspectrometrie.
Verloop van de ziekte
Ernstige, neonatale vorm
Kinderen met de neonatale vorm van propionacidemie worden meestal zonder problemen geboren. Enkele dagen na de geboorte worden ze door ontregeling van de stofwisseling echter ziek en gaan achteruit. Er treedt een verzuring op van het bloed en door het propionzuur stijgen ook andere schadelijke stoffen zoals ammoniak. Dit alles veroorzaakt dat de baby slecht drinkt, suf tot comateus wordt, abnormale bewegingen vertoont, en epileptische aanvallen kan krijgen. Er is risico op blijvende hersenschade of overlijden. In de opvolging kan, ondanks goede behandeling, een aantasting van de hartspier ontwikkelen.
Later begin, presentatie meer dan 1 maand na de geboorte
Sommige patiënten raken onder normale omstandigheden niet ontregeld door het eiwit in de voeding. Zij kunnen echter wel ontregeld raken op momenten dat hun lichaam extra energie nodig heeft, zoals bij infecties of bij te weinig voedselinname. Zo’n ontregeling kan net zo ernstig zijn als bij patiënten met de neonatale vorm. Daarom worden ook deze patiënten na de diagnose behandeld met een eiwitbeperkt dieet, om verdere ontregelingen zoveel mogelijk te voorkomen.
Behandeling
Dit dieet is vergelijkbaar met dat voor MMA. De behandeling bestaat typisch uit een dieet waarin natuurlijk voedingseiwit beperkt wordt, aangevuld met supplementen via een aminozuurmengsel (zonder valine en isoleucine). Daarnaast zijn supplementen L-carnitine nodig. Het vitamine biotine kan soms een gunstig effect hebben.
Dit dieet zal samen worden opgesteld met de metabole arts en diëtist. Het is sterk afhankelijk van de bloedwaarden. Het advies is om dit dieet het hele leven aan te houden.
Bij ziekte en verminderde voedselinname wordt het dieet aangepast (verdere vermindering van eiwit en verhoging van koolhydraten), en soms kan opname voor toediening van een infuus nodig zijn.
Propionacidemie is erfelijk
Propionacidemie is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met propionacidemie drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
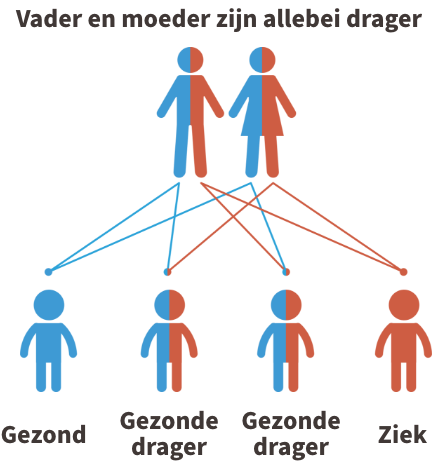
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopies, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien jullie allebei drager zijn, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. U kunt steeds meer informatie krijgen over erfelijkheidsadvies bij de Centra voor menselijke erfelijkheid.