
MADD is een zeldzame erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en afbraak van stoffen in ons lichaam bedoeld. De afbraak van vetten in ons lichaam gebeurt in verschillende stappen. Voor elke stap is er een ander eiwit (enzym) nodig. De afbraak van vetten is belangrijk voor de energievoorziening (“brandstof”) van ons lichaam in niet-gevoede omstandigheden.
MADD wordt veroorzaakt door een defect in het electronentransfer flavoproteine (EFT) of EFT-dehydrogenase. Deze eiwitten staan in voor de overdracht van energie van de verbranding van vetzuren naar de ademhalingsketen van de cel. In de ademhalingsketen van de cel wordt deze energie omgezet zodat die kan gebruikt worden voor allerhande taken van de cel.
Er worden drie vormen van de ziekte onderscheiden:
- een vorm geassocieerd met aangeboren afwijkingen
- een vorm die tot uiting komt bij pasgeborenen maar zonder aangeboren afwijkingen
- een vorm die tot uiting komt na de eerste levensmaand met een te laag bloedsuikergehalte (hypoglycemie) of spierafbraak (rhabdomyolyse)
In tegenstelling tot MCADD is dit een zeer zeldzame stofwisselingsziekte met een voorkomen van minder dan 1:100.000 pasgeborenen in Vlaanderen.
Diagnose
De diagnose wordt gesteld op het acylcarnitineprofiel op gedroogd bloed en bevestigd door DNA-analyse. Eventueel kan de restactiviteit van het enzym gemeten worden in witte bloedcellen, maar meestal op fibroblasten (stukje huid dient afgenomen te worden).
Het acylcarnitineprofiel in een gedroogd bloedspotje geeft de diagnose in stabiele omstandigheden in de neonatale periode, en tijdens decompensatie op latere leeftijd. De techniek die hiervoor aangewend wordt is de tandem-massaspectrometrie. De diagnose kan ook gesteld worden op profiel van organische zuren in de urine.
Verloop van de ziekte
Bij de vorm die geassocieerd is met aangeboren afwijkingen kunnen de klachten al snel na de geboorte aanwezig zijn. Het gaat dan om vaak prematuur geboren zuigelingen met ernstige spierzwakte, hypoglycemie, leverstoornissen en een nieraandoening. Ook worden bij dit type aangeboren afwijkingen (o.a. ter hoogte van de hersenen) gevonden. Vaak overlijden deze baby’s binnen enkele weken of maanden.
Bij de vorm die tot uiting komt bij pasgeborenen maar zonder aangeboren afwijkingen treden de problemen ook binnen een week na de geboorte op. Hier is er meestal sprake van cardiomyopathie (hartspierzwakte) die een ernstig/fataal verloop kan hebben.
Bij de derde vorm kunnen de eerste ziekteverschijnselen na weken, maanden of zelfs jaren optreden. Deze ziekteverschijnselen bestaan vooral uit hypoglycemieën en aanvallen van spierafbraak.
MADD veroorzaakt vaak een intellectuele handicap en neurologische afwijkingen. Onbehandeld is er een slechte prognose wat betreft de levensverwachting.
Behandeling
Goed gespreide voeding met een hoger aandeel aan koolhydraten, al dan niet met eiwitbeperking, is meestal aangewezen als behandeling. Dit dieet zal samen worden opgesteld met de metabole arts en diëtist. Het is sterk afhankelijk van de bloedwaarden. Het advies is om dit dieet het hele leven aan te houden. Sommige kinderen reageren gunstig op supplementen van vitamine B2 (riboflavine). Bij de ernstige vormen met hartaantasting kan behandeling met supplementen van ketonlichamen (vetzuurverbrandingsproducten die als brandstof door o.a. de hartspier kunnen gebruikt worden) in één van gespecialiseerde centra voor erfelijke metabole aandoeningen voor verbetering zorgen.
MADD is erfelijk
MADD is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met MADD drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
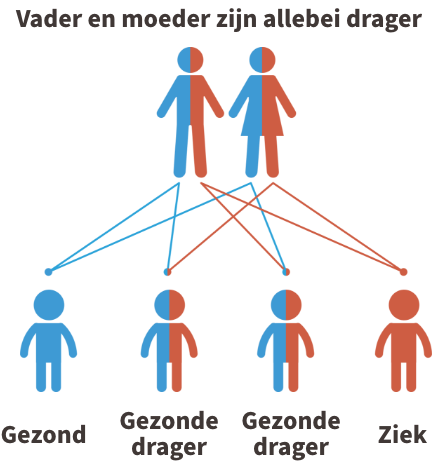
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopies, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien jullie allebei drager zijn, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. U kunt steeds meer informatie krijgen over erfelijkheidsadvies bij de Centra voor menselijke erfelijkheid.