
MCAD deficiëntie is een zeldzame erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en afbraak van stoffen in ons lichaam bedoeld. De afbraak van vetten in ons lichaam gebeurt in verschillende stappen. Voor elke stap is er een ander eiwit (enzym) nodig. De afbraak van vetten is belangrijk voor de energievoorziening (“brandstof”) van ons lichaam in niet-gevoede omstandigheden. Midden lange keten vetten komen zeldzaam in de voeding voor (bv kokosnootolie), maar worden gevormd tijdens de afbraak van lange keten vetten, die wel normaal in de voeding zitten.
Bij MCAD deficiëntie verloopt de afbraak van midden lange keten vetten niet goed door een slechte werking van het enzym medium-chain acyl-CoA dehydrogenase. Dit leidt tot een tekort aan ‘brandstof’ wanneer het lichaam dat juist nodig heeft, zoals bij slecht eten, koorts of bij sporten. Als gevolg daarvan kan er leverschade optreden en kan het bloedsuikergehalte gevaarlijk laag worden.
MCAD deficiëntie hoeft geen klachten te geven. Er zijn mensen die nooit hebben gemerkt dat zij MCAD deficiëntie hebben. MCAD deficiëntie is een goed te behandelen ziekte en met een goede, evenwichtige, voeding en vermijden van vasten is de levensverwachting normaal. Daarom is het van belang de ziekte vroeg op te sporen.
MCAD deficiëntie komt in Vlaanderen bij ongeveer 1:14.000 pasgeborenen voor.
Diagnose
De diagnose wordt gesteld op het acylcarnitineprofiel op gedroogd bloed en bevestigd door DNA-analyse. Eventueel kan de restactiviteit van het enzym gemeten worden in witte bloedcellen.
Het acylcarnitineprofiel in een gedroogd bloedspotje geeft de diagnose in stabiele omstandigheden in de neonatale periode, en tijdens ontregelde stofwisseling op latere leeftijd. De techniek die hiervoor aangewend wordt is de tandem-massaspectrometrie.
Verloop van de ziekte
Een kind met MCAD deficiëntie lijkt gezond bij de geboorte. Bij slecht eten wordt het kind echter slap en suf en kan in coma raken door te weinig suiker in het bloed (hypoglycemie) en leverschade. Dit gebeurt bij onschuldige infecties en onvoldoende eten of vasten. Een klein aantal kinderen is reeds ziek op het moment van de hielprik.
Vetzuren zijn een energiebron die voornamelijk belangrijk zijn wanneer de koolhydraatreserve in de lever opgebruikt is. Bij te lang vasten leidt MCAD deficiëntie tot een ontregelde stofwisseling met levensbedreigende bloedsuikertekorten en leverschade. Bij acute ontregeling met hypoglycemie is er een belangrijk risico op onomkeerbare neurologische schade (epilepsie, verlammingsverschijnselen, gedrag- en ontwikkelingsstoornissen) of overlijden.
Behandeling
Een goede voeding voorkomt klachten. In de eerste levensmaanden wordt het kind ook ’s nachts gevoed. Zoals ieder kind, kan een kind met MCAD deficiëntie ziek worden en daardoor minder goed eten of drinken, waardoor ontregeling van de stofwisseling kan ontstaan. Het dieet dient dan ook aangepast te worden of het kind dient in een ziekenhuis te worden opgenomen voor een infuus. Het is belangrijk om (vooral) de eerste levensjaren door een aangepast dieet de bloedsuiker op pijl te houden door aanvoer van suiker via de voeding. Het kind wordt hierin begeleid door een arts en diëtist.
De meeste mensen met (ernstige vorm van) MCAD deficiëntie krijgen een tekort aan ‘vrij carnitine’, een stof die het lichaam nodig heeft voor het verbranden van vetten. Dit tekort wordt tegengegaan door het toedienen innemen van extra L-carnitine.
MCAD Deficiëntie is erfelijk
MCAD deficiëntie is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met MCAD deficiëntie drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
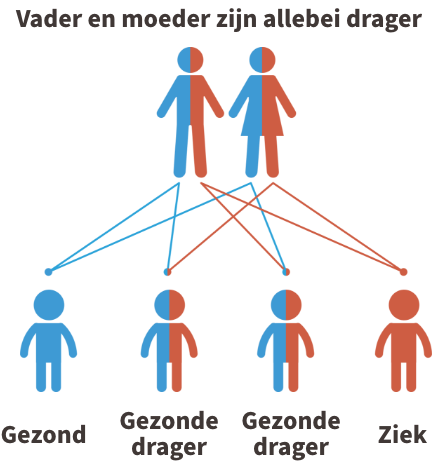
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopies, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien jullie allebei drager zijn, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. Je kan steeds meer informatie krijgen over erfelijkheidsadvies bij de Centra voor menselijke erfelijkheid.