
Glutaaracidemie type 1 is een zeldzame erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en de afbraak van stoffen in ons lichaam bedoeld. Deze aanmaak en afbraak verlopen in verschillende aparte stappen. Voor elke stap is er meestal een specifiek enzym dat ervoor zorgt dat de stap kan doorgaan. Als zo’n enzym niet goed werkt, biochemische afwijking, blokkeert de stofwisseling op die stap, wat aanleiding kan geven tot een stofwisselingsziekte.
Bij glutaaracidemie type 1 worden de aminozuren (de bouwstenen van eiwitten) lysine en tryptofaan niet goed afgebroken. Dat komt door een defect in het enzym glutaryl-CoA dehydrogenase. Lysine en tryptofaan zitten in eiwitten in de voeding (onder andere in vlees, vis en zuivelproducten). Het defect in het enzym glutaryl-CoA dehydrogenase leidt tot een teveel aan glutaarzuur in het lichaam. Deze stof is schadelijk, vooral voor de hersenen.
Glutaaracidurie type 1 komt ongeveer bij 1 op de 90.000 pasgeborenen voor in Vlaanderen.
Diagnose
De diagnose wordt gesteld op het acylcarnitineprofiel met verhoging van C5-DC op gedroogd bloed en bevestigd door DNA-analyse. Eventueel kan de restactiviteit van het enzym gemeten worden in witte bloedcellen.
Het acylcarnitineprofiel in een gedroogd bloedspotje geeft de diagnose in stabiele omstandigheden in de neonatale periode, en tijdens decompensatie op latere leeftijd. De techniek die hiervoor aangewend wordt is de tandem-massaspectrometrie.
Verloop van de ziekte
Een kind met glutaaracidurie type 1 lijkt gezond. Echter, vanaf de geboorte is er sprake van een groot hoofd. Zonder behandeling ontstaat meestal in een periode van slecht eten en koorts een sterke verhoging van het glutaarzuur door de afbraak van de eigen eiwitten. We spreken dan van een metabole crisis.
Tijdens een metabole crisis kan het kind suf worden, stuipen krijgen en kan ernstige hersenschade optreden. Door de hersenschade ontstaan onvrijwillige bewegingen met gestoorde spierspanning. Afhankelijk van hoe ernstig de hersenschade is, kan ook een intellectuele handicap ontstaan, en sommige kinderen overlijden tijdens een metabole crisis.
Eens de hersenschade er is kan deze niet meer ongedaan gemaakt worden. De onvrijwillige bewegingen en intellectuele handicap blijven dus aanwezig.
Behandeling
Glutaaracidurie type 1 is zeer goed te behandelen en de levensverwachting is met een dieet vrijwel normaal. Daarom is het van belang om de ziekte zo snel mogelijk op te sporen zodat de behandeling kan gestart worden voor de eerste metabole crisis.
Het dieet bestaat uit een beperking in natuurlijke eiwitten met aminozuursupplementen aangepast aan de aandoening met vooral een beperking van lysine en tryptofaan. Supplementen van L-carnitine geven bijkomende bescherming tegen metabole crisissen en neurologische verwikkelingen.
Glutaaracidurie-1 is erfelijk
Glutaaracidurie type 1 is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met glutaaracidurie type 1 drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
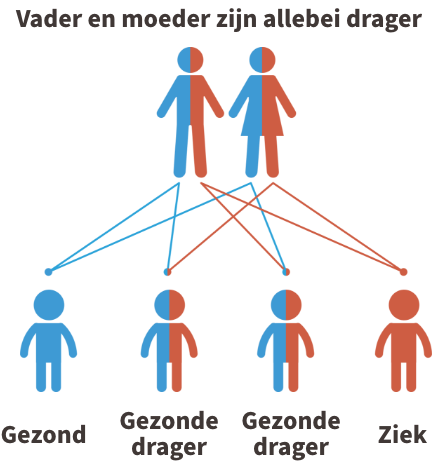
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopies, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien je allebei drager bent, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. Je kunt steeds meer informatie krijgen over erfelijksheidsadvies bij de Centra voor menselijke erfelijkheid.