
Voorkomen in onze populatie
De prevalentie van carnitine palmitoyltransferase type I (CPT I) deficiëntie wordt geschat op < 1:1.000.000 pasgeborenen in Vlaanderen.
Verloop van de ziekte
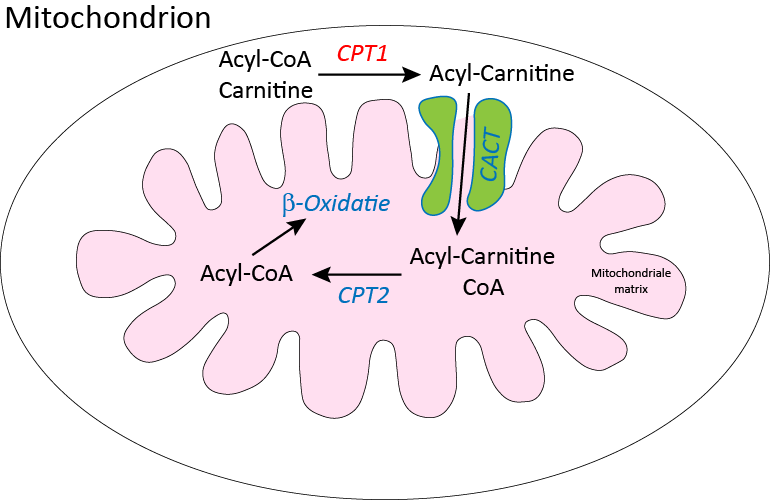
CPT I is verantwoordelijk voor de eerste stap van de carnitine cyclus. Bij de carnitinecyclus worden vetzuren met lange keten eerst gebonden aan carnitine door CPT I. Vervolgens wordt het vetzuur in de tweede stap getransporteerd over de binnenste mitochondriale membraan naar de mitochondriale matrix. In de matrix wordt het vetzuur in de derde stap losgemaakt van carnitine en gekoppeld aan coenzyme A waarna het geoxideerd kan worden en zo energie levert aan de cel. Met de afbraakproducten van vetzuren kunnen in de lever ook ketonlichamen gemaakt worden.
Ketonlichamen kunnen in delen van het lichaam die geen vetzuren afbreken, zoals de hersenen, gebruikt worden als alternatieve energiebron in plaats van glucose.
CPT I deficiëntie wordt veroorzaakt door een genetisch defect in het CPT1A gen. Hierdoor werkt de carnitinecyclus niet goed en kunnen lange keten vetzuren niet goed gebruikt worden als energiebron. Dit veroorzaakt vooral problemen bij periodes van ziekte of vasten, situaties die bij een normaal metabolisme aanleiding geven tot afbraak van vetten als alternatieve energiebron voor glucose. De klinische verschijnselen treden meestal op voor de leeftijd van 2 jaar.
De ziekte uit zich in omstandigheden van infectie of vasten door een hypoglycemie zonder ketose, geassocieerd aan leverdysfunctie en soms ook gestegen spierenzymen, nierproblemen, hartproblemen en hyperammoniëmie. Hierbij is er risico op hersenbeschadiging met intellectuele en motorische handicap en zelfs overlijden als mogelijk gevolg. In sommige gevallen ontwikkelt het kind door leverfalen een hepatische encefalopathie of Reye-like syndroom.
Deze ziekte kan aanleiding geven tot overlijden of irreversibele hersenschade tijdens de eerste decompensatie.
Vroegtijdige diagnose door neonatale screening en behandeling voorkomt alle gevolgen van de ziekte.
Opsporing en diagnostiek
Neonatale screening gebeurt d.m.v. tandem massaspectrometrie (Tandem MS). ESI-MSMS wordt sinds 2007 in de neonatale screening in Vlaanderen aangewend voor de opsporing van de aminozurenafwijkingen PKU en MSUD, de organische acidemieën MMA/PA-IVA en GA1 en de mitochondriale vetzuuroxidatiestoornissen MCADD en MADD.
Opsporen van CPT I deficiëntie gebeurt via vaststellen van een afwijkend acylcarnitine-profiel in het gedroogd bloedstaal (verhoogd C0 of vrij carnitine en een verhoogde ratio van C0/C16+C18). C0 wordt bij elke analyse meegenomen als kwaliteitsparameter bij de screening via ESI-MSMS. De ratio C0/C16+C18 is een specifieke parameter voor de opsporing van CPT I deficiëntie.
De definitieve diagnose wordt gesteld en bevestigd door enzymdiagnostiek op lymfocyten of fibroblasten en/of mutatie-analyse van het CPT1A-gen. Enzymdiagnostiek op fibroblasten is aangewezen bij onduidelijke resultaten van mutatie-analyse.
Behandeling en opvolging
Verdere diagnostiek, behandeling en opvolging van opgespoorde patiëntjes dient te gebeuren in één van de Gespecialiseerde Centra voor zeldzame monogenische erfelijke metabole ziekten (referentiecentra), waar een multidisciplinaire aanpak mogelijk is met o.a. gespecialiseerde diëtisten.
De behandeling van CPT-1A berust hoofdzakelijk op de preventie van katabolisme bij ziekte door snel aanleggen van een glucose-infuus, vergelijkbaar met het beleid bij MCADD. De prognose is goed wanneer decompensaties zo vermeden worden.
Regelmatige voedselinname is gewenst. Periodes van vasten moeten vermeden worden. Nachtelijke sondevoeding en inname van maïszetmeel voor het slapengaan kunnen nodig zijn.
Verder berusten preventieve maatregelen op het volgen van een dieet (beperken van lange keten vetzuren en suppletie door middel van middellange keten vetzuren).
In acute situaties wordt een hoge dosis glucose toegediend via infuus. Indien nodig wordt insuline toegediend,de glucose-toevoer wordt nooit afgebouwd wanneer hyperglycemie optreedt.
Argumenten voor bevolkingsonderzoek naar deze aandoening
Vroege opsporing van CPT I deficiëntie voorkomt decompensatie en de gevolgen daarvan door preventieve maatregelen mogelijk te maken (te vergelijken met MCADD). De neonatale screening op gedroogde bloedspots met behulp van tandem MS is een betrouwbare, praktisch haalbare en betaalbare test, die tevens als aanvaardbaar voor de ouders ervaren wordt. Er zijn bijkomende testen beschikbaar voor het stellen van de definitieve diagnose van de aandoening.
Genetica
CPT I deficiëntie wordt veroorzaakt door mutaties in het CPT1A gen. Het betreft een autosomaal recessief overerfbare aandoening (herhalingsrisico van 1:4 binnen het gezin bij elke zwangerschap).