
Homocystinurie (cystathionine-β-synthase deficiëntie) is een zeldzame aangeboren erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en de afbraak van chemische stoffen in ons lichaam bedoeld. Deze aanmaak en afbraak verlopen in verschillende aparte stappen. Voor elke stap is er meestal een specifiek enzym dat ervoor zorgt dat de stap kan doorgaan. Als zo’n enzym niet goed werkt, biochemische afwijking, blokkeert de stofwisseling op die stap, wat aanleiding kan geven tot een stofwisselingsziekte. Het deel van de stofwisseling dat verstoord is bij homocystinurie betreft eiwitten en hun bouwstenen, de aminozuren.
Bij homocystinurie is er een afwijking in het cystathionine-β-synthase. Cystathionine-β-synthase is nodig voor de afbraak van homocysteïne. Die afbraak is nodig omdat homocysteïne schadelijk is, maar ook om in verdere stappen cysteïne te maken, een ander aminozuur. Homocysteïne ontstaat uit methionine, een aminozuur, wanneer methionine gebruikt wordt in bepaalde belangrijke chemische reacties (methyleringsreacties) in het lichaam. Als cystathionine-β-synthase defect is, treedt er een schadelijke toename op van homocysteïne. Daarnaast is er een tekort aan cysteïne. Deze biochemische afwijkingen treden pas op na de geboorte. Kinderen met homocystinurie zijn normaal bij de geboorte.
Vooral de hersenen, de ogen, het skelet en de bloedvaten zijn kwetsbaar voor de gevolgen van homocystinurie.
Homocystinurie is vrijwel steeds zeer goed behandelbaar door een combinatie van dieet, voedingssupplementen en medicatie (betaïne). Hoe vroeger de behandeling gestart kan worden, hoe beter irreversibele schade aan hersenen, ogen, skelet en bloedvaten voorkomen kan worden.
Homocystinurie komt voor bij ongeveer 1/200.000 pasgeborenen.
Diagnose
Deze ziekte wordt opgespoord door verschillende stoffen of metabolieten te meten in het bloedkaartje. Het gaat om methionine (meer bepaald de verhouding methionine/fenylalanine) en homocysteïne. De diagnose wordt daarna bevestigd met een nieuwe bloedname, een urinestaal, en een DNA-analyse van het CBS-gen, dat de genetische code bevat van het defecte cystathionine-β-synthase.
Verloop van de ziekte
Baby’s met homocystinurie zien er normaal uit bij de geboorte. Vaak is er een vertraagde verstandelijke ontwikkeling. Oogafwijkingen, met name een uitgesproken bijziendheid en een verplaatsing van de ooglens treden vaak op na kleuterleeftijd. Deze oogafwijkingen, waarvoor oogchirurgie nodig is (loslating en verplaatsing van de ooglenzen), zijn vaak de aanleiding tot verder onderzoek naar homocystinurie en de uiteindelijke diagnose. Vaak hebben deze kinderen bij het ouder worden opvallend lange armen en benen en een grote gestalte. Ook andere skeletafwijkingen, onder andere van de borstkas, komen vaak voor (trechterborst). Het gestegen homocysteïne verhoogt bovendien in belangrijke mate het risico op vorming van bloedklonters, zowel in slagaders (arteries) als in aders (venen). Hierdoor treden er vaak op jonge leeftijd beroertes op of nierfalen, maar ook andere organen kunnen hierdoor getroffen worden. Er wordt onderscheid gemaakt tussen kinderen bij wie toedienen van pyridoxine (vitamine B6), een cofactor van cystathionine-β-synthase, de biochemische afwijkingen deels of volledig corrigeert, en kinderen bij wie er geen duidelijk effect is van toediening van pyridoxine. Deze laatste groep wordt behandeld met een dieet en medicatie (zie onder behandeling).
Met vroegtijdige behandeling, vooraleer zichtbare tekens van de ziekte aanwezig zijn, ontwikkelt de overgrote meerderheid van deze kinderen zich normaal.
Behandeling
De behandeling van homocystinurie gebeurt door een team van zorgverleners in de Gespecialiseerde Centra voor monogenische erfelijke metabole aandoeningen. Er worden vitamines zoals pyridoxine, foliumzuur en vitamine B12 gegeven. Bij de kinderen die niet gunstig reageren op pyridoxine is ook een eiwitarm dieet met aminozuursupplementen zonder methionine noodzakelijk. Bijkomend is betaïne meestal nodig om de omzetting van homocysteïne terug naar methionine te bevorderen, en zo homocysteïne verder te doen dalen. Deze combinatiebehandeling voorkomt vrijwel steeds de gevolgen van de ziekte indien de behandeling gestart wordt voor het optreden van zichtbare tekens van de ziekte. Andere vitamines en mineralen, naast suppletie met het aminozuur L-cysteine, worden toegediend naargelang de behoeften volgens de leeftijd. Een strikte opvolging is nodig om een goede controle te verzekeren.
Homocystinurie is erfelijk
Homocystinurie is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met homocystinurie drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
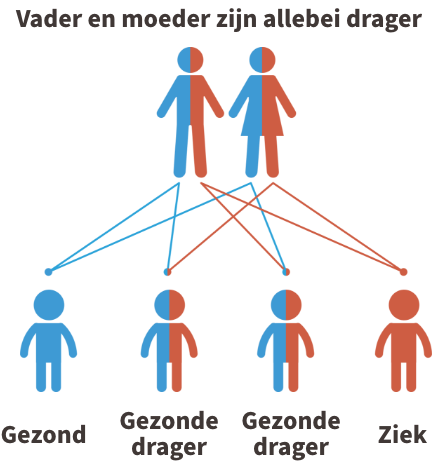
Ouders zijn dragers
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van homocystinurie zijn niet ziek.
Krijgt het kind echter twee afwijkende kopieën, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien beide ouders drager zijn, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. Je kan steeds meer informatie krijgen over erfelijkheidsadvies bij de Centra voor menselijke erfelijkheid.