
Holocarboxylase synthetase deficiëntie (HLCS deficiëntie) is een zeldzame aangeboren erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en de afbraak van chemische stoffen in ons lichaam bedoeld. Deze aanmaak en afbraak verlopen in verschillende aparte stappen. Voor elke stap is er meestal een specifiek enzyme dat ervoor zorgt dat de stap kan doorgaan. Als zo’n enzyme niet goed werkt blokkeert de stofwisseling op die stap, wat aanleiding kan geven tot een stofwisselingsziekte.
Bij HLCS deficiëntie is er een afwijking in het holocarboxylase synthetase. Holocarboxylase synthetase zorgt ervoor dat biotine, een vitamine, op de juiste manier wordt ingebouwd in een reeks van enzymen, de carboxylasen. Deze carboxylasen spelen vooral een rol in de stofwisseling van eiwitten, suiker en vetten. Het inbouwen van biotine in de carboxylasen is noodzakelijk om deze enzymen te activeren. Bij HLCS deficiëntie wordt biotine niet correct ingebouwd in de carboxylasen, en werken de carboxylasen daardoor niet. Dat zorgt ervoor dat verschillende aanmaak- en afbraakprocessen in het lichaam die afhankelijk zijn van werkende carboxylasen, niet kunnen gebeuren. Als gevolg daarvan treedt er een schadelijke toename op van verschillende stoffen. Deze toename treedt pas op na de geboorte. Kinderen met HLCS deficiëntie zijn dus normaal bij de geboorte.
Vooral de hersenen en de huid zijn kwetsbaar voor de toename van schadelijke stoffen bij HLCS deficiëntie.
HLCS deficiëntie is vrijwel steeds zeer goed behandelbaar door toedienen van hoge dosissen van biotine. Hoe vroeger de behandeling gestart kan worden, hoe beter hersenschade voorkomen kan worden.
Diagnose
Deze ziekte wordt opgespoord door verschillende stoffen te meten in het bloedkaartje. Meer bepaald gaat het om C5-OH carnitine, methylcitraat en hydroxypropionzuur. De diagnose wordt daarna bevestigd met een nieuwe bloedname, een urinestaal, en een DNA-analyse van het HLCS-gen, dat de genetische code bevat van het defecte holocarboxylase synthetase.
Verloop van de ziekte
Baby’s met HLCS deficiëntie zien er normaal uit bij de geboorte. Als de ziekte niet opgespoord wordt treden de problemen meestal op in de loop van de eerste 5 levensmaanden. Zonder behandeling treedt bij kinderen met HLCS deficiëntie vaak een acute levensbedreigende ontregeling van de stofwisseling op. Deze ontregeling gaat gepaard met sufheid, braken, epilepsie (stuipen), verzuring van het bloed en verhoogd ammoniak. Zonder behandeling treden ook huidafwijkingen en haaruitval op en hebben deze kinderen meestal een intellectuele handicap. Met vroegtijdige behandeling, vooraleer zichtbare tekens van de ziekte aanwezig zijn, ontwikkelt de overgrote meerderheid van deze kinderen zich normaal.
Behandeling
De behandeling van HLCS deficiëntie gebeurt door een team van zorgverleners in de Gespecialiseerde Centra voor monogenische erfelijke metabole aandoeningen. Er wordt biotine, een vitamine, gegeven in hoge dosis. Deze behandeling voorkomt vrijwel steeds de gevolgen van de ziekte indien de behandeling gestart wordt voor het optreden van zichtbare tekens van de ziekte. Een strikte opvolging is nodig om een goede controle van de stofwisseling te verzekeren.
HLCS deficiëntie is erfelijk
HLCS deficiëntie is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met HLCS deficiëntie drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder wordt dit uitgebeeld.
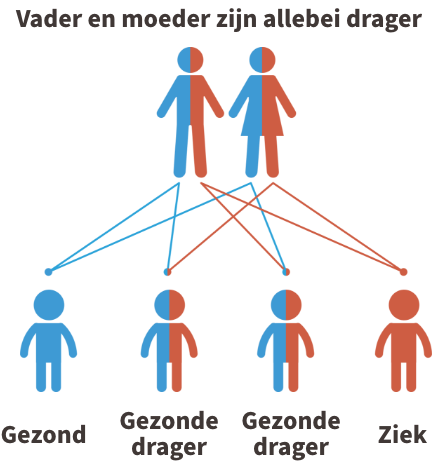
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopies, dus eentje van de vader en eentje van de moeder, dan zal het kindje de ziekte krijgen. Er is bij elke zwangerschap een risico van 1 op 4 dat een kind de twee afwijkende kopies overerft. Je kunt steeds meer informatie krijgen over erfelijksheidsadvies bij de Centra voor menselijke erfelijkheid.