
CPT1 deficiëntie is een zeer zeldzame erfelijke stofwisselingsziekte. Met ‘stofwisseling’ wordt de aanmaak en afbraak van stoffen in ons lichaam bedoeld. Vetzuren zijn een soort vetten die een keten bevatten van variabele lengte en zijn een belangrijke bron van energie. De afbraak van vetzuren in ons lichaam gebeurt in verschillende stappen. Voor elke stap is er een ander eiwit (enzym) nodig Zo verloopt het transport van lange keten vetzuren naar de plaats waar ze kunnen afgebroken worden in 3 stappen. De eerste stap in dit transport wordt vervuld door CPT1, en bestaat uit het koppelen van het vetzuur aan carnitine. De afbraak van vetzuren is belangrijk voor de energievoorziening (“brandstof”) van ons lichaam in niet-gevoede omstandigheden. Bij niet-eten (vasten) komen de lange keten vetzuren vrij uit de vetopslag van het eigen lichaam.
Bij CPT1 deficiëntie verloopt de afbraak van lange keten vetzuren, vanuit de voeding of de vetopslag, niet goed door een slechte werking van CPT I. Dit leidt tot een tekort aan ‘brandstof’ wanneer het lichaam dat juist nodig heeft, zoals bij slecht eten, koorts of bij sporten. Het gevolg is een daling van het bloedsuikergehalte (hypoglycemie) die gevaarlijk is.
De eerste symptomen worden vaak zichtbaar tussen de leeftijd van 0-18 maanden. Het verloop kan zeer ernstig zijn met, naast daling van het bloedsuikergehalte, ook leveraantasting, en zelfs overlijden. Dit kan voorkomen worden door de vroege opsporing via de neonatale screening (‘hielprik’) en goede opvolging.
CPT1 deficiëntie is een goed te behandelen ziekte en met een goede, evenwichtige, voeding en vermijden van vasten is de levensverwachting normaal. Daarom is het belangrijk de ziekte vroeg op te sporen. Het is een zeer zeldzame ziekte maar goed te behandelen zodat besloten werd om ze op te nemen in het screeningsprogramma van Vlaanderen.
CPT1 komt in Vlaanderen vermoedelijk voor bij minder dan 1/1.000.000 pasgeborenen (precieze gegevens zullen duidelijker worden door de neonatale screening in Vlaanderen over meerdere jaren te evalueren).
Diagnose
De diagnose wordt gesteld op gedroogd bloed met tandem-massaspectrometrie. Met dit onderzoek wordt, door zowel vrij carnitine als carnitine gebonden aan vetzuren te meten, een zogenaamd “acylcarnitineprofiel” bepaald. Als CPT1 niet werkt wordt er een sterke toename van vrij carnitine vastgesteld. Deze afwijking van het acylcarnitineprofiel is duidelijk bij ziekte of vasten, maar ook wanneer de baby in een goede stabiele toestand is. Dit laat toe de ziekte op te sporen bij de hielprik vooraleer er problemen optreden. DNA-analyse bevestigt de diagnose. Eventueel kan de activiteit van CPT I gemeten worden in witte bloedcellen of huidcellen.
Verloop van de ziekte
Een kind met CPT1 deficiëntie lijkt gezond. Vetten zijn een energiebron die voornamelijk belangrijk is bij vasten. Bij slecht eten of infecties wordt het kind slap en suf en kan het in coma raken of overlijden door een tekort aan suiker in het bloed (hypoglycemie). Het ammoniak in het bloed kan ook verhoogd zijn. Dit treedt op bij een metabole decompensatie. Deze metabole decompensaties kunnen aanleiding geven tot onomkeerbare hersenschade (epilepsie, motorische handicap, intellectuele handicap, gedragsstoornissen).
Behandeling
De behandeling van CPT1 deficiëntie gebeurt door een team van zorgverleners in de Gespecialiseerde Centra voor monogenische erfelijke metabole aandoeningen. Een goede voeding met vermijden van vasten voorkomt klachten. Op jonge leeftijd zal het nodig zijn om het kind ook ’s nachts te voeden. Zoals ieder kind kan een kind met CPT1 deficiëntie ziek worden en daardoor niet goed eten of drinken, waardoor metabole decompensatie kan ontstaan. Het dieet moet dan aangepast worden of het kind moet in een ziekenhuis opgenomen worden voor een infuus. Het is belangrijk om de eerste 7 levensjaren door een aangepast dieet de bloedsuiker op pijl te houden door verhoogde aanvoer van suiker via de voeding.
CPT1 deficiëntie is erfelijk
CPT1 deficiëntie is een autosomaal recessieve aandoening. Dit betekent dat beide ouders van een kindje met CPT I deficiëntie drager zijn van de ziekte (of de ziekte zelf hebben). In de figuur hieronder aan wordt dit uitgebeeld.
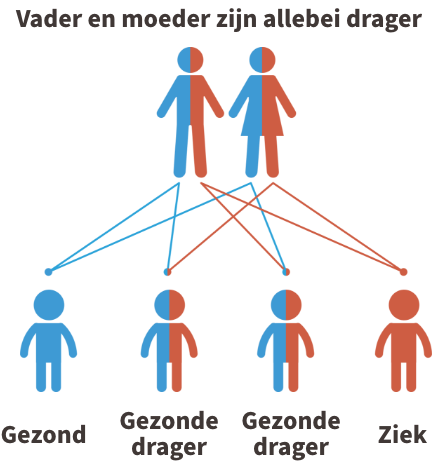
Kinderen krijgen vrijwel alle erfelijke eigenschappen in tweevoud, namelijk een kopie van de moeder en een kopie van de vader. Iemand die 1 afwijkende kopie heeft gekregen van de vader of de moeder, wordt een drager genoemd. Dragers van de ziekte zijn nooit ziek.
Krijgt het kind echter twee afwijkende kopieën, dus eentje van de vader en eentje van de moeder, dan is het kindje ziek.
Indien jullie allebei drager zijn, dan is er 1 kans op 4 dat het kindje de ziekte zal hebben. Je kan steeds meer informatie krijgen over erfelijkheidsadvies bij de Centra voor menselijke erfelijkheid.